Sample-basierte Krylov-Quantendiagonalisierung von'm fermionischen Gittermodell
Schätzung vom Rechenaufwand: Neun Sekunden auf'm Heron-r2-Prozessor (HINWEIS: Das ist nur eine Schätzung. Die tatsächliche Laufzeit kann variieren.)
Hintergrund
In dem Tutorial zeigen wir, wie ma sample-basierte Quantendiagonalisierung (SQD) verwendet, um die Grundzustandsenergie von'm fermionischen Gittermodell zu schätzen. Konkret untersuchen wir das eindimensionale Single-Impurity-Anderson-Modell (SIAM), das zur Beschreibung von magnetischen Verunreinigungen in Metallen verwendet wird.
Das Tutorial folgt einem ähnlichen Workflow wie das verwandte Tutorial Sample-basierte Quantendiagonalisierung von'm Chemie-Hamiltonian. Der wesentliche Unterschied liegt aber darin, wie die Quantenschaltkreise aufgebaut san. Das andere Tutorial verwendet einen heuristischen Variationsansatz, der für Chemie-Hamiltonians mit potenziell Millionen von Wechselwirkungstermen attraktiv ist. Dieses Tutorial hingegen verwendet Schaltkreise, die Zeitentwicklung durch den Hamiltonian approximieren. Solche Schaltkreise können tief sein, was diesen Ansatz besser für Anwendungen auf Gittermodelle geeignet macht. Die von diesen Schaltkreisen vorbereiteten Zustandsvektoren bilden die Basis für einen Krylov-Unterraum, und daher konvergiert der Algorithmus nachweislich und effizient zum Grundzustand, unter geeigneten Annahmen.
Der in diesem Tutorial verwendete Ansatz lässt sich als Kombination der in SQD und Krylov-Quantendiagonalisierung (KQD) verwendeten Techniken betrachten. Der kombinierte Ansatz wird manchmal als sample-basierte Krylov-Quantendiagonalisierung (SQKD) bezeichnet. Schau dir Krylov-Quantendiagonalisierung von Gitter-Hamiltonians für ein Tutorial zur KQD-Methode an.
Dieses Tutorial basiert auf der Arbeit „Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", auf die du für weitere Details verweisen kannst.
Single-Impurity-Anderson-Modell (SIAM)
Der eindimensionale SIAM-Hamiltonian ist eine Summe aus drei Termen:
wobei
Dabei san die fermionischen Erzeugungs- und Vernichtungsoperatoren für die Bad-Stelle mit Spin , san die Erzeugungs- und Vernichtungsoperatoren für den Verunreinigungsmodus, und . , und san reelle Zahlen, die die Hüpf-, Onsite- und Hybridisierungswechselwirkungen beschreiben, und ist eine reelle Zahl, die das chemische Potential angibt.
Zu beachten ist, dass der Hamiltonian ein spezieller Fall des allgemeinen wechselwirkenden Elektronen-Hamiltonians ist:
wobei aus Einteilchen-Termen besteht, die quadratisch in den fermionischen Erzeugungs- und Vernichtungsoperatoren san, und aus Zweiteilchen-Termen, die quartisch san. Für das SIAM gilt:
und enthält die restlichen Terme im Hamiltonian. Um den Hamiltonian programmatisch darzustellen, speichern wir die Matrix und den Tensor .
Orts- und Impulsbasis
Aufgrund der näherungsweisen Translationssymmetrie in erwarten wir nicht, dass der Grundzustand in der Ortsbasis dünn besetzt ist (die Orbitalbasis, in der der Hamiltonian oben angegeben ist). Die Leistung von SQD ist nur dann garantiert, wenn der Grundzustand dünn besetzt ist, das heißt, er hat signifikantes Gewicht nur auf einer kleinen Anzahl von Rechenbasiszuständen. Um die Dünnbesetztheit des Grundzustands zu verbessern, führen wir die Simulation in der Orbitalbasis durch, in der diagonal ist. Diese Basis nennen wir die Impulsbasis. Da ein quadratischer fermionischer Hamiltonian ist, kann er effizient durch eine Orbitalrotation diagonalisiert werden.
Näherungsweise Zeitentwicklung durch den Hamiltonian
Zur Approximation der Zeitentwicklung durch den Hamiltonian verwenden wir eine Trotter-Suzuki-Zerlegung zweiter Ordnung:
Unter der Jordan-Wigner-Transformation entspricht die Zeitentwicklung durch einem einzelnen CPhase-Gate zwischen dem Spin-up- und dem Spin-down-Orbital an der Verunreinigungsstelle. Da ein quadratischer fermionischer Hamiltonian ist, entspricht die Zeitentwicklung durch einer Orbitalrotation.
Die Krylov-Basiszustände , wobei die Dimension des Krylov-Unterraums ist, werden durch wiederholte Anwendung eines einzelnen Trotter-Schritts gebildet:
Im folgenden SQD-basierten Workflow sampeln wir aus dieser Menge von Schaltkreisen und verarbeiten die kombinierte Menge von Bitstrings mit SQD nach. Dieser Ansatz unterscheidet sich von dem im verwandten Tutorial Sample-basierte Quantendiagonalisierung von'm Chemie-Hamiltonian verwendeten, wo Samples aus einem einzigen heuristischen Variationsschaltkreis gezogen wurden.
Voraussetzungen
Bevor du mit diesem Tutorial anfängst, stell sicher, dass du Folgendes installiert hast:
- Qiskit SDK v1.0 oder neuer, mit Unterstützung für Visualisierung
- Qiskit Runtime v0.22 oder neuer (
pip install qiskit-ibm-runtime) - SQD Qiskit Addon v0.11 oder neuer (
pip install qiskit-addon-sqd) - ffsim (
pip install ffsim)
Schritt 1: Problem auf'n Quantenschaltkreis abbilden
Zuerst erzeugen wir den SIAM-Hamiltonian in der Ortsbasis. Der Hamiltonian wird durch die Matrix und den Tensor dargestellt. Dann rotieren wir ihn in die Impulsbasis. In der Ortsbasis platzieren wir die Verunreinigung an der ersten Stelle. Wenn wir aber in die Impulsbasis rotieren, verschieben wir die Verunreinigung an eine zentrale Stelle, um Wechselwirkungen mit anderen Orbitalen zu erleichtern.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 20
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
Als nächstes erzeugen wir die Schaltkreise zur Erzeugung der Krylov-Basiszustände. Für jede Spinspezies ist der Anfangszustand durch die Überlagerung aller möglichen Anregungen der drei Elektronen, die dem Fermi-Niveau am nächsten san, in die 4 nächsten leeren Moden ausgehend vom Zustand gegeben, und wird durch die Anwendung von sieben XXPlusYYGates realisiert. Die zeitentwickelten Zustände werden durch aufeinanderfolgende Anwendungen eines Trotter-Schritts zweiter Ordnung erzeugt.
Für eine detailliertere Beschreibung dieses Modells und wie die Schaltkreise entworfen san, schau dir „Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization" an.
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Schritt 2: Problem für die Quantenausführung optimieren
Jetzt, wo wir die Schaltkreise erstellt haben, können wir sie für eine Zielhardware optimieren. Wir wählen den am wenigsten ausgelasteten QPU mit mindestens 127 Qubits. Schau dir die Qiskit IBM® Runtime-Dokumentation für mehr Informationen an.
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
Using backend ibm_fez
Jetzt verwenden wir Qiskit, um die Schaltkreise für das Ziel-Backend zu transpilieren.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Schritt 3: Mit Qiskit-Primitiven ausführen
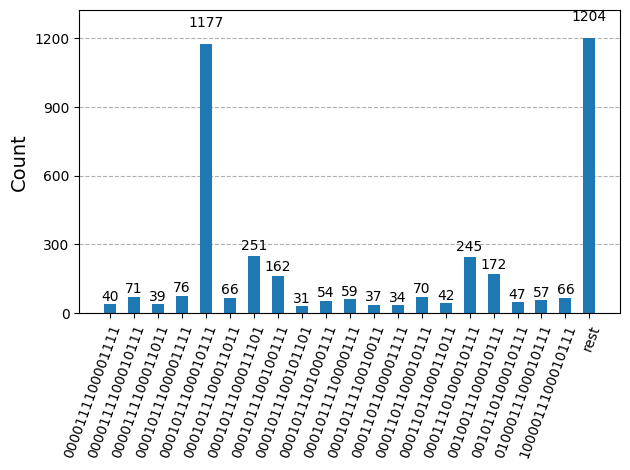
Nachdem wir die Schaltkreise für die Hardwareausführung optimiert haben, sind wir bereit, sie auf der Zielhardware laufen zu lassen und Samples für die Grundzustandsenergieabschätzung zu sammeln. Nachdem wir das Sampler-Primitiv verwendet haben, um Bitstrings aus jedem Schaltkreis zu sampeln, kombinieren wir alle Ergebnisse in einem einzigen Counts-Dictionary und plotten die 20 am häufigsten gesampelten Bitstrings.
from qiskit.visualization import plot_histogram
from qiskit_ibm_runtime import SamplerV2 as Sampler
# Sample from the circuits
sampler = Sampler(backend)
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the counts from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
Schritt 4: Nachbearbeiten und Ergebnis ins gewünschte klassische Format zurückgeben
Jetzt führen wir den SQD-Algorithmus mit der Funktion diagonalize_fermionic_hamiltonian aus. Schau dir die API-Dokumentation für Erklärungen zu den Argumenten dieser Funktion an.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -28.61321893815165
Subspace dimension: 10609
Subsample 1
Energy: -28.628985564542244
Subspace dimension: 13924
Subsample 2
Energy: -28.620151775558114
Subspace dimension: 10404
Iteration 2
Subsample 0
Energy: -28.656893066053115
Subspace dimension: 34225
Subsample 1
Energy: -28.65277622004119
Subspace dimension: 38416
Subsample 2
Energy: -28.670856034959165
Subspace dimension: 39601
Iteration 3
Subsample 0
Energy: -28.684787675404362
Subspace dimension: 42436
Subsample 1
Energy: -28.676984757118426
Subspace dimension: 50176
Subsample 2
Energy: -28.671581704249885
Subspace dimension: 40804
Iteration 4
Subsample 0
Energy: -28.6859683054753
Subspace dimension: 47961
Subsample 1
Energy: -28.69418206537316
Subspace dimension: 51529
Subsample 2
Energy: -28.686083516445752
Subspace dimension: 51529
Iteration 5
Subsample 0
Energy: -28.694665630711178
Subspace dimension: 50625
Subsample 1
Energy: -28.69505984237118
Subspace dimension: 47524
Subsample 2
Energy: -28.6942873883992
Subspace dimension: 48841
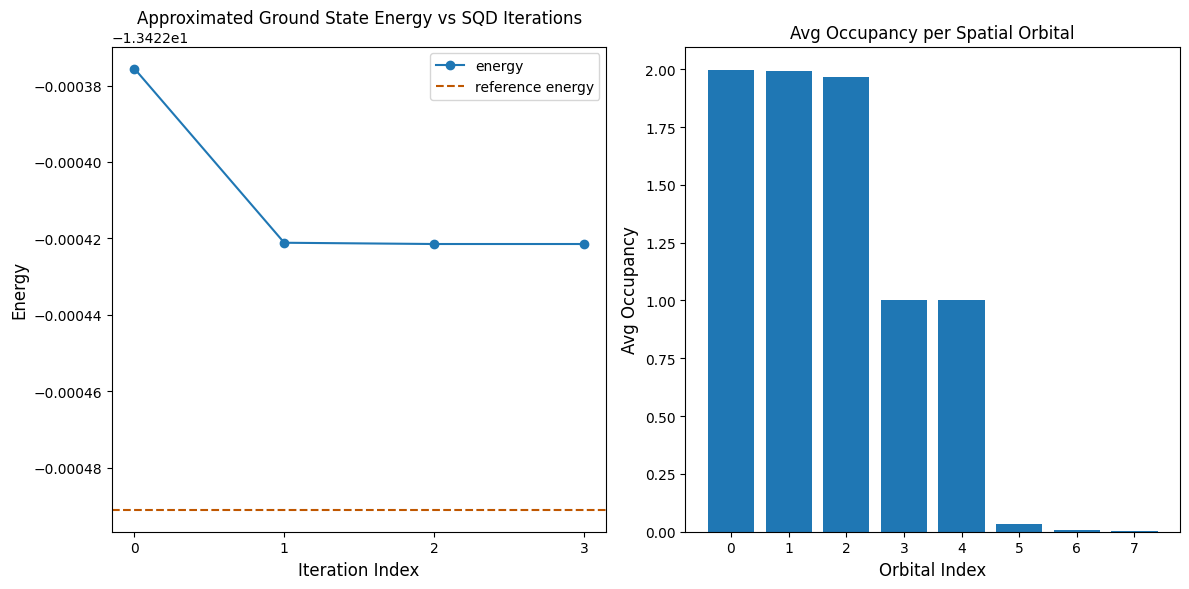
Die folgende Code-Zelle plottet die Ergebnisse. Das erste Diagramm zeigt die berechnete Energie als Funktion der Anzahl der Konfigurationswiederherstellungs-Iterationen, und das zweite Diagramm zeigt die durchschnittliche Besetzung jedes räumlichen Orbitals nach der letzten Iteration. Als Referenzenergie verwenden wir die Ergebnisse einer DMRG-Berechnung, die separat durchgeführt wurde.
import matplotlib.pyplot as plt
dmrg_energy = -28.70659686
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=dmrg_energy, color="#BF5700", linestyle="--", label="DMRG energy"
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference (DMRG) energy: {dmrg_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - dmrg_energy):.5f}")
plt.tight_layout()
plt.show()
Reference (DMRG) energy: -28.70660
SQD energy: -28.69506
Absolute error: 0.01154
Energie verifizieren
Die von SQD zurückgegebene Energie ist garantiert eine obere Schranke für die wahre Grundzustandsenergie. Der Wert der Energie kann verifiziert werden, weil SQD auch die Koeffizienten des Zustandsvektors zurückgibt, der den Grundzustand approximiert. Du kannst die Energie aus dem Zustandsvektor mithilfe seiner 1- und 2-Teilchen-reduzierten Dichtematrizen berechnen, wie in der folgenden Code-Zelle gezeigt.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -28.69506